
THEORETICAL STUDY OF NEW
CANDIDATE ORGANIC MATERIALS FOR PHOTOVOLTAIC APPLICATIONS
Anass EL Karkri 1![]()
![]() ,
Imane EL Mhamedi 1
,
Imane EL Mhamedi 1![]() , Zakaria EL Malki 1
, Zakaria EL Malki 1![]() , Mohammed Bouachrine 2
, Mohammed Bouachrine 2![]()
1 High
School of Technology, Moulay Ismaïl University, Meknes,
Morocco
2 Higher
School of Technology-EST Khenifra, Sultan Moulay Sliman University, Benimellal,
Morocco
|
|
|
ABSTRACT |
|
|
Our work
consists of a theoretical prediction, through DFT and TD-DFT methods, of the
electronic and optical properties of six conjugated organic compounds used as
electron donor materials in BHJ solar cells, of which PCBM is the acceptor
material. This study is necessary to discuss the effect of substituents
(donor units) on the different properties of these compounds, and to predict
promising materials in organic solar cells using the AMPS-1D simulation
software. The results obtained show that all the molecules have good
geometric, electronic, and optical properties, thus showing an increase in
the power conversion efficiency of photovoltaic cells based on these
materials, which reaches a value of 17% for the molecule P-Eth-TEdotT-A. |
|||
|
Received 08 March 2023 Accepted 09 April
2023 Published 26 April 2023 Corresponding Author Anass EL Karkri anass.elkarkri@gmail.com DOI 10.29121/IJOEST.v7.i2.2023.485 Funding: This research
received no specific grant from any funding agency in the public, commercial,
or not-for-profit sectors. Copyright: © 2023 The
Author(s). This work is licensed under a Creative Commons
Attribution 4.0 International License. With the
license CC-BY, authors retain the copyright, allowing anyone to download,
reuse, re-print, modify, distribute, and/or copy their contribution. The work
must be properly attributed to its author.
|
|||
|
Keywords: Organic Material,
DFT, Solar Cell, Power Conversion Efficiency |
|||
1. INTRODUCTION
Since their discovery, organic materials based on conjugated molecules have aroused continuous interest due to their relevance in several fields of application, such as batteries Yuan and Wang (2020), light-emitting diodes Amin, et al. (2019), transistors Guo et al. (2021) and photovoltaic cells Riaz et al. (2020). Many researchers have been interested in the synthesis of short-chain compounds based on conjugated molecules because they are not amorphous and can be synthesized as well-defined structures Wegner and Klaus (2008). Molecules or conjugated polymers containing thiophene fragments have attracted much attention due to their unique electronic properties, high photoluminescence quantum efficiency and thermal stability Zhang et al. (2019).
These properties depend on the degree of electronic delocalization in these materials and the modification of the chemical structure due to the incorporation of charge carriers. In order to obtain materials having a predominant capacity, the development of new structures is currently undertaken using molecular engineering.
In this context, quantum chemical methods have been increasingly used to predict the gap energy of conjugated systems Zgou et al. (2008), Bouzzine et al. (2008), Scharber et al. (2021) and for the choice of suitable materials to optimize the properties of organic photovoltaic devices Malki et al. (2018), Wang et al. (2018), Malki et al. (2017), Malki et al. (2020), Karkri et al. (2020).
Compared to traditional silicon solar cells where free charges are generated directly upon photoexcitation, exciton separation in BHJ organic solar cells requires overcoming a larger potential barrier (exciton binding energy > 50 meV) by due to the lower dielectric constant of the organic/polymer medium Coropceanu et al. (2007). In BHJ solar cells this is usually achieved by mixing materials with different electron affinities and oxidation potentials, i.e., an electron donor is used in conjunction with an electron acceptor, such than a PCBM or fullerene, which has molecular orbital energy states that facilitate electron transfer Nielsen et al. (2015).
Figure 1
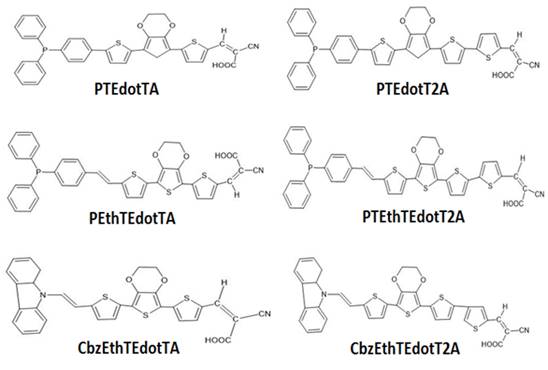
|
Figure 1 Chemical Structure of the Studied Molecules Mi (I=1-6) |
This work presents the results of a theoretical study on electron donor groups, the aim of which was to identify potential candidates for use in organic photovoltaic cells. The calculations are made using the Gaussian 09 software, with the functional B3LYP and the basis 6-31 G (d, p). More specifically, it is a study of the effect of substitutions of atoms and donor units in these systems to understand how these substitutions could control certain properties of the molecules. In particular, we sought to optimize properties such as the optical gap, the energy of the HOMO (Highest Occupied Molecular Orbital) and the energy of the LUMO (Lowest Unoccupied Molecular Orbital) of the compounds with the aim of optimizing them for use in photovoltaic cells. In general, the ideal optical gap for a solar cell is around 1.3 eV, and the HOMO and LUMO frontier orbitals of materials must be calibrated to form a BHJ (Bulk Heterojunction) junction with the electron acceptor like the (6,6) methyl phenyl-C61-butyrate denoted PCBM. However, theoretical calculations are made on six Thiophene-based electron donor materials (Figure 1) to determine their geometric, electronic, and optical properties, and also the study of the photovoltaic properties of solar cells based on these materials, and the impact of the variation of the active layer thickness and temperature on their performance using the AMPS-1D simulation software.
2. MATERIALS AND METHODS
Density functional theory (DFT) using the exchange and correlation functional B3LYP Roquet et al. (2006) and Pople's basis 6-31G (d,p) Mikroyannidis et al. (2001) was used to determine the geometry and electronic properties of all materials studied. The calculations were carried out with the Gaussian program 09 Frisch et al. (2009). It has been shown that the DFT-B3LYP/6-31G (d, p) level is sufficient to accurately determine the geometric and energetic properties, the electronic structure, the absorption, and emission spectra of many π-conjugates organic molecules Ditchfield et al. (1971). The energies of the orbitals HOMO, LUMO and the gap (EHOMO-ELUMO) are also deduced from the most stable conformations of the molecules studied in their ground state. Excited state energies and oscillator strengths (OS) were calculated by the TD/DFT method Casida (1995) from the fully optimized ground state structure. According to the results obtained, the UV-Visible absorption and emission spectra were simulated using the GaussView 4.1.2 software Dennington et al. (2007). In addition, the representation of the orbital density of the HOMO and the LUMO was carried out at the same level of theory using the program GaussView. Berny's algorithm using redundant internal coordinates Peng et al. (1996) was employed for energy minimization.
3. RESULTS AND DISCUSSIONS
3.1. Optimized structures and geometric properties
The
optimized ground state geometries (S0) of the six molecules studied in this
work are shown in
Figure 2
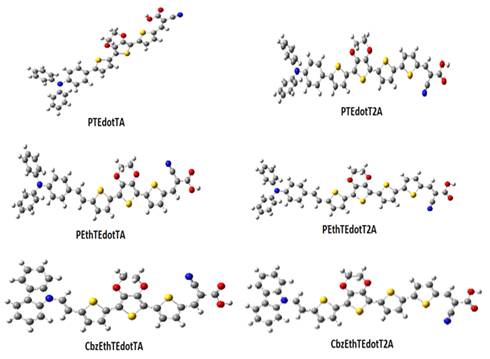
|
Figure 2 Optimized Structures at the B3LYP/6-31G (D, P) Level of the Compounds Studied. |
Figure 2. The most relevant geometric parameters (intercyclic bond lengths d and dihedral angles Φ between adjacent fragments) are reported in Table 1.
Table 1
|
Table 1 Intercyclic Distances (Å) and Dihedral Angle (°) Values of the Studied Compounds Obtained from Neutral State of Optimized Structures Calculated by B3LYP/6-31G (D, P) Level of DFT |
||||||
|
Molécules |
||||||
|
M1: Ptedotta |
P-T |
T-E |
E-T |
T-A |
||
|
D |
1.46239 |
1.43722 |
1.43196 |
1.42637 |
||
|
F |
23.30 |
-0.843 |
1.195 |
-0.0329 |
||
|
M2: Ptedott2a |
P-T |
T-E |
E-T |
T-T |
T-A |
|
|
D |
1.46289 |
1.43792 |
1.43393 |
1.43630 |
1.42194 |
|
|
F |
24.423 |
0.663 |
1.126 |
0.926 |
0.151 |
|
|
M3: Pethtedotta |
(P-Th)-T |
T-E |
E-T |
T-A |
||
|
D |
1.44031 |
1.43528 |
1.43066 |
1.42124 |
||
|
F |
-0.2206 |
-0.258 |
0.923 |
-0.235 |
||
|
M4: Pethtedott2a |
(P-Eth)-T |
T-E |
E-T |
T-T |
T-A |
|
|
D |
1.44037 |
1.43582 |
1.43351 |
1.43624 |
1.42195 |
|
|
F |
-0.707 |
-0.354 |
-0;381 |
-0.119 |
0.012 |
|
|
M5: Cbzethtedotta |
(Cbz-Eth)-T |
T-E |
E-T |
T-A |
||
|
D |
1.44333 |
1.43589 |
1.43111 |
1.42162 |
||
|
F |
7.95 |
0.287 |
-0.586 |
0;28 |
||
|
M6: Cbzethtedott2a |
(Cbz-Eth)-T |
T-Edot |
Edot-T |
T-T |
T-A |
|
|
D |
1.44336 |
1.43639 |
1.43392 |
1.43658 |
1.42222 |
|
|
F |
7.65 |
0.158 |
-0.853 |
0.827 |
-0.00658 |
|
The structural properties of a material that can be used in a photovoltaic device are very important. In fact, the more this material adopts a planar geometry, the more the intramolecular charge transfer is favorable. From Table 1, the analysis of the values of the dihedral angles Φ shows that all the molecules have a similar planar conformation. As for the values of the bond lengths, they are about 1.420 – 1.463 Å. Indeed, in these D-π-A systems, the π-conjugated group is used as an intramolecular charge transfer (ICT) bridge. Thus, the bond length between the D group and the π-conjugate can elucidate a real interaction in the system. This short bond distance can promote intramolecular charge transfer.
3.2. Electronic propertie
Theoretical knowledge of the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is crucial in the study of organic solar cells. Moreover, the HOMO and LUMO energy levels of the donor and the acceptor for photovoltaic devices are very important factors in determining whether the charge transfer between the donor and the acceptor is efficient. These energy levels can be obtained experimentally from the oxidation and reduction potentials measured by cyclic voltammetry Kini et al. (2020). Theoretically, these electronic properties can be determined by the DFT method Mancuso et al. (2020). Therefore, we calculated from the most stable conformation of the molecules studied, the values of the energies HOMO (eV), LUMO (eV) and the gap (eV) at the level B3LYP/6-31G (d, p). The results obtained are grouped in Table 2. The energy levels of the HOMO and LUMO orbitals of organic materials (M 1 to M 6) are presented in Figure 3.
Figure 3
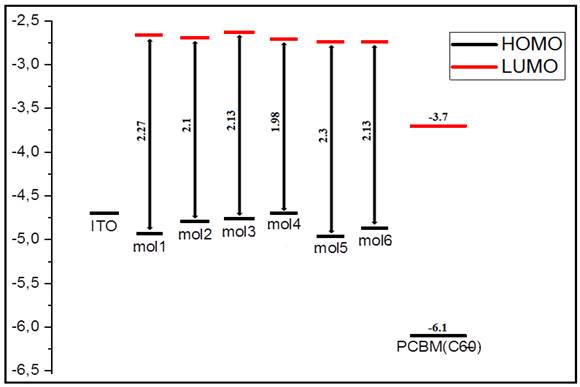
|
Figure 3 Sketch of Calculated Energy of the HOMO and LUMO Levels of Studied Molecules and the Acceptor PCBM |
By analyzing the results reported in this figure, we first notice that the LUMO energy level of all the compounds studied is above that of the acceptor compound (PCBM). The difference between these two levels is of the order of 1.0 eV, thus showing that the transfer of photo-excited charge from M i compounds to PCBM is possible Ghosekar et al (2021).
The ability to donate electrons by the donor group in D-π-A compounds tends to influence the electrochemical properties. A D-π-A compound with a stronger electron donor is expected to give a high HOMO compared to that with a weaker electron donor. Analysis of the results obtained (Figure 3) shows that the energy levels of the HOMOs of these molecules are in the following order, M4 > M3 > M2 > M6 > M1 > M5, which indicates that the M 4 with the highest HOMO of -4.70 eV contains the strongest electron donor group.
The energy gap (Egap) for PTEdotTA, PTEdotT2A, PTEthTEdotTA, PTEthTEdotT2A, (cbz-eth) EdotTA, (cbz-eth) EdotT2A was obtained by the differences in energy levels HOMO and LUMO (HOMO-LUMO) using B3LYP/6-31G (d,p). The band gaps in the case of these compounds are 2.26 eV, 2.09 eV, 2.13 eV, 1.98 eV, 2.3 eV, 2.12 eV respectively. The band gap is generally low for all the systems studied, since these values are generally between 1.98 and 2.30 eV. The observed decrease in gap is due to the insertion of ethylyne, the insertion of thiophene units at the position adjacent to the donor and the insertion of Edot unit at the position adjacent to the acceptor, resulting in an increase in the conjugation length of the molecules, promising better intramolecular charge transfer.
Therefore, all the studied molecules can be used as electron donors because the process of injection of electrons from the excited molecule to the acceptor conduction band (PCBM) and subsequent regeneration is possible in the BHJ Organic solar cell.
Open Circuit Voltage Voc
The Voc parameter of a BHJ solar cell is theoretically related to the energy difference between the molecular orbital (HOMO) of the donor and the LUMO of the acceptor, taking into account the energy loss during charge generation Gadisa et al. (2004). From where, the maximum value of the Voc is estimated according to the following expression:
Voc =
ELUMO (accepteur) - EHOMO (donneur) - 0,3 (1)
Indeed, the higher this difference, the more favorable
the charge transfer.
Table 2 groups the values of Voc
and the electronic affinities EA for all the molecules, calculated by the
relation Anafcheh et al. (2012).
EA= E(M) – E(M-) (2)
Where, E(M) and E(M-) are the total energies of neutral molecule and anion states, respectively.
Mi compounds have high EA values (Table 2), so they are qualified as excellent electron-carrying material.
From the estimated values of Voc
recorded in Table 2, we can notice that they are between 1.00
and 1.26 eV and decrease in the following order: M5 > M1 > M6 > M2
> M3 > M4. Thus, M1 and M5 are the molecules which present the highest
Voc. Therefore, we can suggest that they are good candidates for photovoltaic
applications.
Table 2
|
Table 2 Energy Values of LUMO, HOMO, the
Gap, Voc, EA, of the Studied Molecule Calculated by
B3LYP/6-31G (D, P) Level |
||||||
|
Compounds |
M1 |
M2 |
M3 |
M4 |
M5 |
M6 |
|
EHOMO (eV) |
-4.93 |
-4.79 |
-4.76 |
-4.70 |
-4.96 |
-4.87 |
|
ELUMO (eV) |
-2.66 |
-2.69 |
-2.63 |
-2.71 |
-2.67 |
-2.74 |
|
Egap (eV) |
2.27 |
2.09 |
2.13 |
1.98 |
2.29 |
2.13 |
|
EA (eV) |
1.71 |
1.77 |
1.73 |
1.83 |
1.71 |
1.83 |
|
VOC (eV) |
1.23 |
1.09 |
1.06 |
1 |
1.26 |
1.17 |
3.3. Frontier molecular orbitals
Analysis of OMF iso-density surfaces is important since it provides
a reasonable qualitative indication of excitation properties and electron-hole
transport capacity. Thus, we have represented in Figure 4 the spatial distribution of the electron
densities of the HOMO and LUMO orbitals of all the compounds studied. According
to this Figure, the surfaces of the HOMO electronic distribution of the studied
molecules present an anti-binding character between two adjacent fragments and
binding inside each unit and they are mainly localized
on the electron donor group and the π-conjugate spacer. As for the LUMOs,
they present a binding character between two adjacent fragments, so that the
singlet states of lower energies correspond to an electronic transition of the
type π-π*. In addition, they are essentially located on the
π-spacer and the acceptor group A (2-cyanoacrylic acid). Therefore,
electronic transitions of all D- π -A compounds, from HOMO to LUMO could
lead to intramolecular charge transfer (ICT) from D to A across the
π-spacer, so that the HOMO transition -LUMO can be classified as a
π-π* TCI. On the other hand, the anchoring group (-CNCOOH) of all the
compounds makes considerable contribution to LUMOs which could lead to strong
binding with the PCBM acceptor thus improving the injection efficiency, and
hence increasing the short circuit current density Jsc.
Figure 4
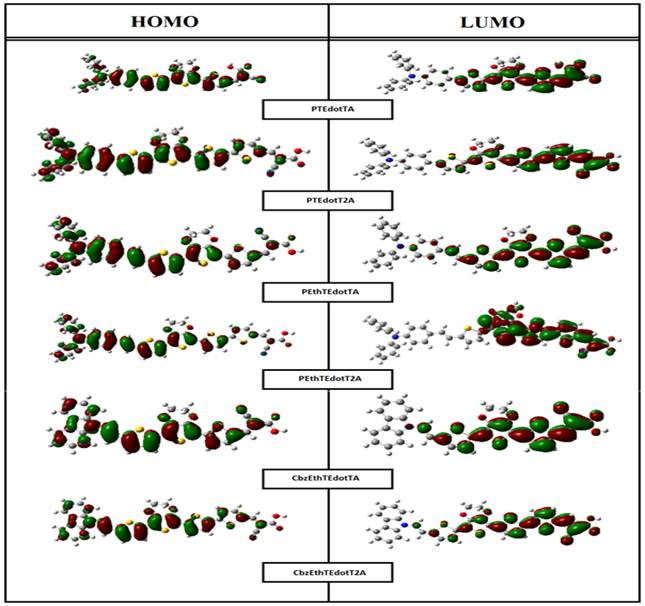
|
Figure 4 The Contour Plots of HOMO and LUMO Orbitals of the Studied Compounds. |
3.4. Optical properties
How the absorption of a new material matches the solar spectrum is
an important factor for application as a photovoltaic material. In addition, a
good photovoltaic material should have visible, broad
and strong absorption characteristics. To better understand the optical
property and electronic transition, the excitation energy and UV-Vis absorption spectra for the singlet-singlet transition of
all molecules were simulated using the TD-DFT method in starting with an
optimized geometry obtained by B3LYP / 6-31G (d, p) level.
The excitation energies of the first singlet transition, the corresponding peak absorption wavelengths and the oscillator strengths of all molecules are listed in Table 3.
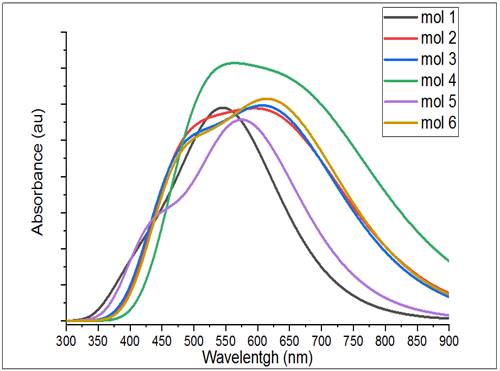
The simulated absorption spectra of the studied compounds obtained at the B3LYP/6-31G (d, p) level are presented in Figure 5.
PTEdotTA shows a series of bands between 399.63
and 458.83nm, and the strongest absorption at 556.97nm belonging to the HOMO -
LUMO transition, PTEdotT2A shows a series of bands between 462.37 and 515.06
nm, and the strongest absorption at 645.66 nm could be attributed to the
electronic transition from HOMO to LUMO. PEthTEdotTA
features a series of bands between 447.92 nm and 492.23 nm, and the absorption
at 634.30 nm is attributed to the electronic transition from HOMO to LUMO.
PEthTEdotT2A features a series of bands between 493.28 nm and 533.26 nm, and
the absorption at 684.32 nm is attributed to the electronic transition from
HOMO to LUMO. CbzEthTEdotTA shows a series of bands
between 405.95 nm and 443.55 nm, and the absorption at 580.86 nm is attributed
to the electronic transition from HOMO to LUMOCbzEthTEdotT2A exhibits a series
of bands between 450.23 nm and 486.35 nm, and the absorption at 635.73 nm is
attributed to the electronic transition from HOMO to LUMO. We noted that the
insertion of the thiophene donor and ethylyne spacer
units into the oligomer backbone affects the electronic structure by donating
charge carriers, thereby lowering the energy band gap
and increasing the conjugation length. The λmax
values of six compounds are in the order of PEthTEdotT2A > PTEdotT2A >
CbzEthTEdotT2A > PEthtTEdotTA > CbzEthTEdotTA > PTEdotTA (see Table 3), which is in excellent agreement with the
corresponding reverse order of Egap values displayed
in the previous section. The oscillator strength (O.S), which corresponds to
the molar extinction coefficient, is also increased by the addition of
supplementary units of thiophene and ethylene. The absorption wavelengths
resulting from the S0 to S1 electronic transition increase gradually with
increasing conjugation lengths.
It is well known that the position (related to the gap between the
HOMO and LUMO levels) and the width of the main band of the absorption spectrum
are the main parameters that can act on the light collection process and
consequently on the efficiency of solar cells. From Figure 5, it clearly appears that the simulated
absorption spectra present a similar profile for all the compounds studied;
they also show an intense main band at high wavelengths from 400 to 850 nm. As
shown in Table 3, the most intense contribution to the main
band is the first singlet excitation HOMO - LUMO.
The absorption spectrum of compounds M1 and M5 shows that they emit
at higher wavelengths (556.97 nm and 580.86 nm) with higher intensity (f1=
1.3356 and f2=1.3339). These encouraging optical properties suggest that these
compounds, as electron donors, will be the best candidates for photovoltaic
devices.
Table 3
|
Table 3 Absorption Spectra Data Obtained by TD-DFT Method for the Mi (I =1–6) Compounds at B3LYP/6-31G (D, P) Optimized Geometries |
|||||
|
Compounds |
Electronic Transition |
λ (nm) |
Eex |
O. S |
MO/Character |
|
M1 |
S0 ⟶
S1 |
556.97 nm |
2.2261 eV |
1.3356 |
HOMO->LUMO (98%) |
|
S0 ⟶
S2 |
458.83 nm |
2.7022 eV |
0.4353 |
H-1->LUMO (97%) |
|
|
S0 ⟶
S3 |
399.63 nm |
3.1025 eV |
0.24 |
H-2->LUMO (33%) |
|
|
M2 |
S0 ⟶
S1 |
645.66 nm |
1.9203 eV |
1.1806 |
HOMO->LUMO (99%) |
|
S0 ⟶
S2 |
515.06 nm |
2.4072 eV |
0.7639 |
H-1->LUMO (96%) |
|
|
S0 ⟶
S3 |
462.37 nm |
2.6815 eV |
0.547 |
HOMO->L+1 (84%) |
|
|
M3 |
S0 ⟶
S1 |
634.30 nm |
1.9547 eV |
1.3097 |
HOMO->LUMO (99%) |
|
S0 ⟶
S2 |
492.23 nm |
2.5189 eV |
0.8926 |
H-1->LUMO (86%) |
|
|
S0 ⟶
S3 |
447.92 nm |
2.7680 eV |
0.262 |
HOMO->L+1 (75%) |
|
|
M4 |
S0 ⟶
S1 |
684.32 nm |
1.8118 eV |
1.311 |
HOMO->LUMO (99%) |
|
S0 ⟶
S2 |
533.26 nm |
2.3250 eV |
1.1392 |
H-1->LUMO (83%) |
|
|
S0 ⟶
S3 |
493.28 nm |
2.5135 eV |
0.4059 |
HOMO->L+1 (75%) |
|
|
M5 |
S0 ⟶
S1 |
580.86 nm |
2.1345 eV |
1.3339 |
HOMO->LUMO (99%) |
|
S0 ⟶
S2 |
443.55 nm |
2.7952 eV |
0.5912 |
H-1->LUMO (72%) |
|
|
S0 ⟶
S3 |
405.95 nm |
3.0542 eV |
0.0724 |
HOMO->L+1 (62%) |
|
|
M6 |
S0 ⟶
S1 |
635.73 nm |
1.9503 eV |
1.3865 |
HOMO->LUMO (99%) |
|
S0 ⟶
S2 |
486.35 nm |
2.5493 eV |
0.9915 |
H-1->LUMO (61%) |
|
|
S0 ⟶
S3 |
450.23 nm |
2.7538 eV |
0.0722 |
HOMO->L+1 (55%) |
|
Figure 5
|
Figure 5 TD-DFT Simulated UV– Visible Optical Absorption Spectra of Studied Compounds |
3.5. Photovoltaic properties
3.5.1. Effect of thickness
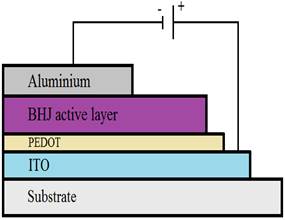
In this part, we will represent a J-V characterization under illumination of the organic cells (whose architecture is represented in Figure 6) based on the materials studied using the numerical simulation software AMPS-1D. This characterization makes it possible to determine several parameters of the cell, such as the open circuit voltage (Voc), the current density (Jsc), the fill factor FF, the PCE and the series and shunt resistances.
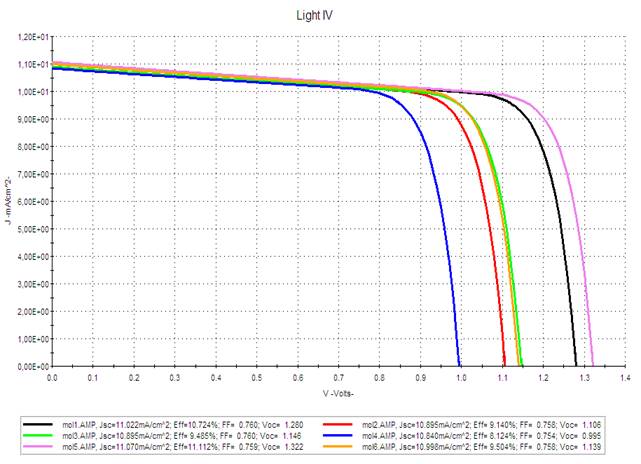
Figure 7 and Figure 8 show the J-V characteristic of the simulated solar cells for active layer thicknesses of 50 and 100 nm respectively. We introduced a layer of PEDOT between the anode and the active layer.
This study allowed us to see the effect of the thickness
of the active layer and the use of different donor materials on the parameters
of the cell.
Figure 6
|
Figure 6 Architecture of the Studied Solar Cell |
It should be noted that the insertion of a layer of PEDOT has an
important role in the collection of charge carriers, because the PEDOT
therefore makes it possible to increase the work function on the ITO side to
5.2 eV Kugler and Salaneck (2000), Brown et al. (1999).
The efficiency 𝛈 is directly
proportional to the Voc delivered by the device.
Furthermore, to have high photoconversion efficiency, a high Voc is required, which is the case for the
molecules M1 and M5 (section 2).
The power conversion efficiency was calculated according to the following equation Chattopadhyay et al. (2002)
![]()
Where
Pin is the incident power density, Jsc
is the short-circuit current, Voc is the
open-circuit voltage, and FF denotes the fill factor.
Figure 7
|
Figure 7 J-V Characteristics for Different Mi For 50 nm Thickness |
Figure 8
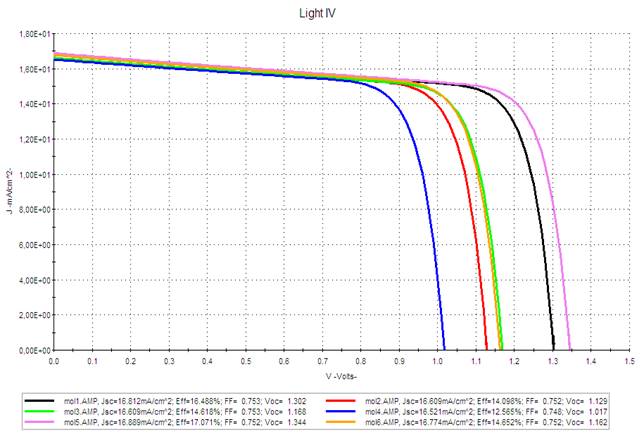
|
Figure 8 J-V Characteristics for Different Mi For 100 nm Thickness |
According to the estimated values of Voc recorded in Table 4, we can notice that they are between 0.995 and 1.322 eV (for a thickness of 100 nm) and decrease in the following order: M5 > M1 > M3 > M6 > M2 > M4. Compounds M1 and M5 occur with the highest Voc. Likewise for the values of the current density Jsc. Thus, the power conversion efficiency of these two molecules has high rates compared to the other molecules, and which further increases by (M1: from 10.724 to 16.488%, and M2: from 11.112 to 17.071%) with the increase in the thickness of the active layer from 50 to 100 nm (Table 4), because enlarging the active layer allows the absorption of more photons, which makes it possible to improve the performance of the studied solar cells.
Therefore, based on
the good results found of Jsc and Voc for M1 and M5 molecules, we can suggest that
they are good candidates for photovoltaic applications.
Table 4
|
Table
4 Parameters
of Solar Cells Based on M1 And M5 as a Function of Temperature |
|||||||
|
Molecules |
Jsc(mA/cm2) |
Voc (V) |
FF |
𝛈 (%) |
Rs (𝛺.cm2) |
Rsh (𝛺.cm2) |
|
|
M1 |
50 nm |
11.022 |
1.280 |
0.760 |
10.724 |
7.10 |
126 |
|
100 nm |
16.812 |
1.302 |
0.753 |
16.488 |
5.34 |
103 |
|
|
M2 |
50 nm |
10.895 |
1.106 |
0.758 |
9.140 |
7.71 |
145 |
|
100 nm |
16.609 |
1.129 |
0.752 |
14.098 |
6.56 |
125 |
|
|
M3 |
50 nm |
10.895 |
1.146 |
0.760 |
9.485 |
7.75 |
153 |
|
100 nm |
16.609 |
1.168 |
0.695 |
14.618 |
7.30 |
146 |
|
|
M4 |
50 nm |
10.840 |
0.995 |
0.754 |
8.124 |
7.52 |
280 |
|
100 nm |
16.521 |
1.017 |
0.748 |
12.565 |
6.25 |
220 |
|
|
M5 |
50 nm |
11.070 |
1.322 |
0.759 |
11.112 |
6.82 |
130 |
|
100 nm |
16.889 |
1.344 |
0.752 |
17.071 |
5.12 |
110 |
|
|
M6 |
50 nm |
10.998 |
1.139 |
0.758 |
9.504 |
7.54 |
218 |
|
100 nm |
16.774 |
1.162 |
0.752 |
14.652 |
6.44 |
185 |
|
|
P3HT-PCBM [32] |
7.58 |
1.07 |
0.628 |
5.094 |
|||
3.5.2. Effect of temperature on the photovoltaic characteristics
In this part, the temperature is varied and the value of the
thickness of the active layer is kept constant at 100 nm. Figure 9 and Figure 10 show the AMPS-1D simulation results of the J
(V) characteristics of the organic solar cell based on M1-PCBM and M5-PCBM
respectively for different temperatures.
Figure 9
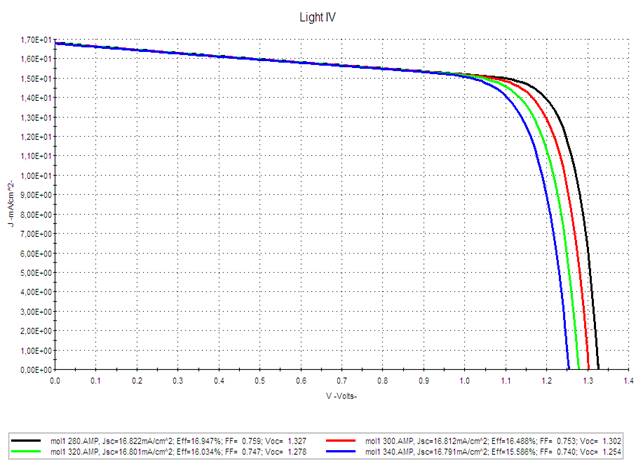
|
Figure 9 AMPS-1D Simulation of the J(V) Characteristics of the M1-PCBM Based Organic Solar Cell for Different Temperatures. |
Figure 10
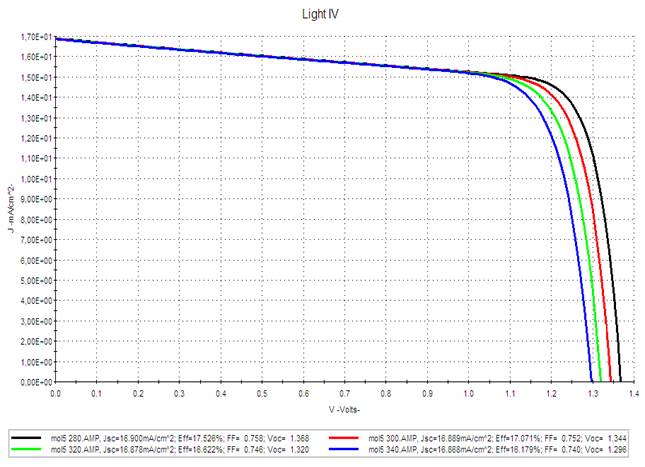
|
Figure 10 AMPS-1D Simulation of the J(V) Characteristics of the M5-PCBM Based Organic Solar Cell for Different Temperatures. |
The results of the simulation are grouped in the table below
Table 5
|
Table 5 Parameters of Solar Cells Based on M1 And M5 as a Function of Temperature. |
|||||||
|
Molecules |
Jsc(mA/cm2) |
Voc (V) |
FF |
𝛈 (%) |
Rs (𝛺.cm2) |
Rsh (𝛺.cm2) |
|
|
280 K |
16.822 |
1.327 |
0.759 |
16.947 |
5.22 |
102 |
|
|
M1 |
300 K |
16.812 |
1.302 |
0.753 |
16.488 |
5.34 |
103 |
|
|
320 K |
16.801 |
1.278 |
0.747 |
320 K |
5.54 |
102 |
|
340 K |
16.791 |
1.254 |
0.740 |
15.588 |
5.63 |
101 |
|
|
280 K |
16.900 |
1.368 |
0.758 |
17.526 |
4.89 |
111 |
|
|
M5 |
300 K |
16.889 |
1.344 |
0.752 |
17.071 |
5.12 |
110 |
|
320 K |
16.878 |
1.320 |
0.748 |
16.622 |
5.58 |
109 |
|
|
340 K |
16.868 |
1.296 |
0.740 |
16.179 |
5.73 |
110 |
|
It seems that
1) The short-circuit current density Jsc
is very insensitive to temperature.
2) The open circuit voltage Voc varies
very slightly with the increase in temperature. These results show that the
optimal functioning of the photovoltaic generator substantially corresponds to
functioning at constant optimal voltage.
3)
The PCE is slightly sensitive to the increase in temperature: when
the temperature increases by 20°C around the ambient temperature and for an
illumination of 100 mW/cm2 the efficiency
decreases by 2.5%.
We
can conclude that increasing the temperature of the organic photovoltaic cell
presents a slight degradation of the photovoltaic parameters.
4. CONCLUSIONS
In this work, the electronic structure, and optical properties of six novel thiophene-based molecules were determined using density functional theory DFT and its time-dependent variant TD-DFT. Through this study, we were able to identify a direct relationship between the power of the donor group and the efficiency of the molecules studied in an organic solar cell. Based on the results obtained, we analyzed the role of the various electron donor groups selected on all the properties sought. Also, we studied their effects on the open circuit voltage Voc, the short circuit current density Jsc, the fill factor FF and the power conversion efficiency of the cell by discussing the key factors affecting these parameters in the aim to find potential compounds to be used in photovoltaics. It can be concluded that these thiophene-based molecules all have good photophysical properties, thus showing good compatibility with PCBM as an acceptor material. We can therefore suggest the potentiality of all the compounds studied in this field. In particular, the M1 and M5 materials were found to be the best oligomers for solar cells, due to their particular properties found and their high values of conversion efficiency (16.488% and 17.071% respectively) comparing with the values found in the literature.
This theoretical approach could be, on the one hand, a good tool for orienting the synthesis of new compounds more useful as active materials in this kind of photovoltaic devices, and on the other hand, to predict the structure-properties relationship of other new compounds for use in this field.
CONFLICT OF INTERESTS
None.
ACKNOWLEDGMENTS
None.
REFERENCES
A. El karkri, Z. El Malki, M. Bouachrine, F.S. Spirau, J.M. (2020). Sotiropoulos. Characterization and Simulation Study of Organic Solar Cells Based on Donor-Acceptor (D-π-A) molecular materials. RSC Adv, 10, 18816. https://doi.org/10.1039/D0RA01815E
A. Gadisa, M. Svensson, M.R. Andersson, O. Inganas (2004). Appl. Phys. Lett. 84, 1609. https://doi.org/10.1063/1.1650878; M.C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A.J. Heeger, C.J. Brabec, Adv. Mater. 18 (2006) 789 https://doi.org/10.1002/adma.200501717; C.J. Brabec, A. Cravino, D. Meissner, N.S. Sariciftci, T. Fromherz, M.T. Rispens, L. Sanchez, J.C. Hummelen, Adv. Funct. Mater. 11 (2001) 374. https://doi.org/10.1063/1.2817930
Anafcheh, M., Ghafouri, R., Hadipour (2012). N.L. Sol. Energy Mater. Sol. Cells, 105, 125. https://doi.org/10.1016/j.solmat.2012.05.015
Brown T. M., Kim J. S., Friend R. H., Cacialli F., Daik R. and Feast W. J. (1999). Built-in field Electro Absorption Spectroscopy of Polymer Light-Emitting Diodes Incorporating a Doped Poly (3,4-Ethylene Dioxythiophene) Hole Injection Layer, Appl. Phys. Lett, 75, 1679- 1681. https://doi.org/10.1063/1.124789
C. Peng, P.Y. Ayala, H.B. Schlegel, M.J. Frisch (1996). Using Redundant Internal Coordinates to Optimize Equilibrium Geometries and Transition States, J. Comput. Chem. 17, 49. https://doi.org/10.1002/(SICI)1096-987X(19960115)17:1
C.B. Nielsen, S. Holliday, H. Chen, S.J. Cryer, I. Mcculloch (2015). Non-fullerene Electron Acceptors for use in Organic Solar Cells Acc. Chem. Res., 48 (11), 2803-2812 https://doi.org/10.1021/acs.accounts.5b00199
Chattopadhyay, D.; Lastella, S.; Kim, S.; Papadimitrakopoulos (2002). Length Separation of Zwitterion-Functionalized Single Wall Carbon Nanotubes by GPC, F. J. Am. Chem. Soc., 124 (5), 728-729. https://doi.org/10.1021/ja0172159
Chen, Yuan, and Chengliang Wang (2020). "Designing High Performance Organic Batteries." Accounts of Chemical Research 53(11), 2636-2647. https://doi.org/10.1021/acs.accounts.0c00465
Coropceanu, J. Cornil, D.A. da Silva Filho, Y. Olivier, R. Silbey, J.-L. Bredas (2007). Charge Transport in Organic Semiconductors Chem. Rev., 107, 926-952. https://doi.org/10.1021/cr050140x
Ghosekar, Ishan C., and Ganesh C. Patil (2021). "Review on Performance Analysis of P3HT: PCBM-based Bulk Heterojunction Organic Solar Cells." Semiconductor Science and Technology 36(4), 045005. https://doi.org/10.1088/1361-6641/abe21b
Guo, Xiaojun, Lei Han, and Xiao Hou (2021). "Insights into the Device Structure, Processing and Material Design for an Organic Thin-Film Transistor Towards Functional Circuit Integration." Materials Chemistry Frontiers 5(18): 6760-6778. https://doi.org/10.1039/D1QM00334H
Hsaine Zgou, Mohamed Hamidi, Jean-Pierre Lére-Porte, Francoise Serein-Spirau, Mohammed Bouachrine (2008). Structural and Electronic Properties of New Materials Based on Thiophene and Phenylene, Acta Physico-Chimica Sinica, 24 (1), 37-40, ISSN 1872-1508, https://doi.org/10.1016/S1872-1508(08)60003-0.
Hussain, Riaz, et al. (2020). "Molecular
engineering of A-D-C-D-A Configured Small Molecular Acceptors (Smas) with
Promising Photovoltaic Properties for High-Efficiency Fullerene-Free Organic
Solar Cells." Optical and Quantum Electronics 52, 1-20.
https://doi.org/10.1007/s11082-020-02482-7
J.A. Mikroyannidis, D.V. Tsagkournos, P. Balraju, G.D. Sharma (2001). Journal of Power Sources, 196, 4152. https://doi.org/10.1016/j.jpowsour.2010.12.038
Kini, Gururaj P., Sung Jae Jeon, and Doo Kyung Moon. (2020)."Design Principles and Synergistic Effects of Chlorination on a Conjugated Backbone for Efficient Organic Photovoltaics: A Critical Review." Advanced Materials 32(11), 1906175. https://doi.org/10.1002/adma.201906175
Kugler T. and Salaneck W. R. (2000). Chemical Species at Polymer/ITO Interfaces: Consequences for the Band Alignment in Light-Emitting Devices, C.R. Acad. Sci., Ser. IV:Phys., Astrophys., 1, 409-423. https://doi.org/10.1016/S1296-2147(00)00140-2
Lu Zhang (2012). Master of Science & Engineering, Science & Engineering, May.
M. Casida (1995). Time Dependent Density Functional Response Theory for Molecules, D.P. Chong (Ed.), Recent Advances in Density Functional Methods, 1 World Scientific, Singapore (1995) 155; M.E. Casida, C. Jamorski, K.C. Casida, D.R. Salahub, J. Chem. Phys., 108 (1998), 4439; R.E. Stratmann, G.E. Scuseria, M.J. Frisch, J. Chem. Phys., 109 (1998) 8218 ; S. Hirata, M. Head-Gordon Chem. Phys. Lett., 302 (1999) 375. https://doi.org/10.1016/S0009-2614(99)00137-2
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, J.A. Montgomery, T. Vreven,
Jr., K.N. Kudin, J.C. Burant,
J.M. Millam, S.S. Iyengar, J. Tomasi, V. Barone, B. Mennucci,
M. Cossi, G. Scalmani, N. Rega, G.A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M.Ishida,
T. Nakajima (2009). 167 Y. Honda, O. Kitao, H.
Nakai, M. Klene, X. Li,
J.E. Knox, H.P. Hratchian, J.B. Cross, C. Adamo, J. Jaramillo, R.Gomperts, R.E. Stratmann, O. Yazyev, A.J.
Austin, R. Cammi, C. Pomelli,
J.W. Ochterski, P.Y. Ayala, K. Morokuma,
G.A. Voth, P. Salvador, J.J. Dannenberg,
V.G. Zakrzewski, S. Dapprich,
A.D. Daniels, M.C. Strain, O. Farkas, D.K. Malick,
A.D. Rabuck, K. Raghavachari,
J.B. Foresman, J.V. Ortiz, Q. Cui, A.G. Baboul, S. Clifford, J. Cioslowski,
B.B. Stefanov, G. Liu, A. Liashenko,
P. Piskorz, I. Komaromi,
R.L. Martin, D.J. Fox, T. Keith, M.A. Al- Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe,
P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, C. Gonzalez, J.A. Pople, GAUSSIAN 03, Revision
B.04, Gaussian, Inc., Pittsburgh PA.
Mancuso, Jenna L., et al. (2020). "Electronic Structure Modeling of Metal-Organic Frameworks." Chemical Reviews 120(16), 8641-8715. https://doi.org/10.1021/acs.chemrev.0c00148
R. Ditchfield, W.J. Hehre, J.A. Pople, J. Chem. Phys., 54 (1971). 76, W.J. Hehre, R. Ditchfiesld, J.A. Pople, J. Chem. Phys., 56 (1972) 643; P.C. Hariharan, J.A. Pople, Mol. Phys., 27 (1974) 209; M.S. Gordon, Chem. Phys. Lett., 76 (1980) 33. https://doi.org/10.1007/978-94-009-8736-4_4
R. I.I. Dennington, T. Keith, J. Millam (2007). GaussView Version 4.1.2, Semichem Inc., Shawnee Mission, KS.
S. Roquet, A. Cravino, P. Leriche, O. Aleveque, P. Frere, J. Roncali. (2006). J. Am. Chem. Soc. 128, 3459.
S.M. Bouzzine, A. Makayssi, M. Hamidi, M. Bouachrine
(2008). Bridging effect on structural and optoelectronic properties of
oligothiophene, Journal of Molecular Structure:
THEOCHEM, 851 (1-3), 254-262, ISSN 0166-1280,
https://doi.org/10.1016/j.theochem.2007.11.023.
Salehi, Amin, et al. (2019)."Recent
Advances in OLED Optical Design." Advanced Functional Materials 29(15),
1808803. https://doi.org/10.1038/s41377-020-0341-9.
Scharber, Markus Clark, and Niyazi Serdar Sariciftci (2021). "Low Band Gap Conjugated Semiconducting Polymers." Advanced Materials Technologies 6(4), 2000857. https://doi.org/10.1002/admt.202000857
Wang, Y., Qian, D., Cui, Y., Zhang, H., Hou, J., Vandewal, K., Kirchartz, T., Gao, F. (2018). Optical Gaps of Organic Solar Cells as a Reference for Comparing Voltage Losses, Adv. Energy Mater. 8, 1801352. https://doi.org/10.1002/aenm.201801352
Wegner, G., and M. Klaus (2008). eds. "Electronic Materials: The Oligomer Approach.
Z. EL Malki, M. Bouachrine, F. Serein-Spirau, JM. Sotiropoulos (2018). International Journals of Advanced Research in Computer Science and Software Engineering ISSN: 2277-128X, 8 (12), 38-51.
Z. EL Malki, M. Bouachrine, L. Bejjit, M. Haddad, F. Serein-Spirau (2017). J-M. Sotiropoulos, International Journals of Advanced Research in Computer Science and Software Engineering ISSN: 2277- 128X , 7 (6), 96-107.
Z. El Malki, A. El karkri, M.
Bouachrine, F.S. Spirau (2020). Springer Nature
Switzerland AG, Theoretical Study of New Compounds (D1-BT-EDOT-BT-D2-A) Based
on 3, 4-Ethylenedioxythiophene (EDOT) and Benzothiadiazole
https://doi.org/10.1007/978-3-030-36475-5_28; (BT) for
Dye Sensitized Solar Cells. AI2SD 2019. Lecture Notes in Electrical
Engineering, 624. Springer, Cham. https://doi.org/10.1007/978-3-030-36475-5_28
Zhang, Yuan‐Lan, et al. (2019).
"High‐efficiency Red Organic Light‐Emitting Diodes With
External Quantum Efficiency Close To 30% Based on a Novel Thermally Activated
Delayed Fluorescence Emitter." Advanced Materials 31(42), 1902368.
https://doi.org/10.1002/adma.201902368
 This work is licensed under a: Creative Commons Attribution 4.0 International License
This work is licensed under a: Creative Commons Attribution 4.0 International License
© Granthaalayah 2014-2023. All Rights Reserved.

